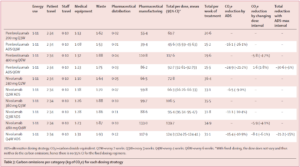
Question évaluée : L’étude vise à démontrer la faisabilité de préparations anticipées d’irinotécan à différentes doses (200, 300, 370 mg) en poches de perfusion de polyoléfine (POF), en évaluant leur stabilité physico-chimique et microbiologique à 4°C et à l’abri de la lumière. Une étude de stabilité en conditions d’utilisation simulées sur 24h à 25°C a également été réalisée.
Type d’étude : Étude de stabilité physico-chimique et microbiologique.
Méthode :
- Poches de 250 mL en polyoléfine contenant de l’irinotécan à 1,6 ou 2,3 mg/mL, dilué dans du NaCl 0,9 %,
- Conservation à +2 et +8 °C (analyses à J0-J3-J7-J14-J21-J28-J35-J42-J49-J56-J63-J73-J77-J84),
- Conditions d’utilisations simulées :
- Conservation à +2 et +8 °C pendant 84 jours puis dosage à 25°C à H0, H8, H10, H24
- Analyses :
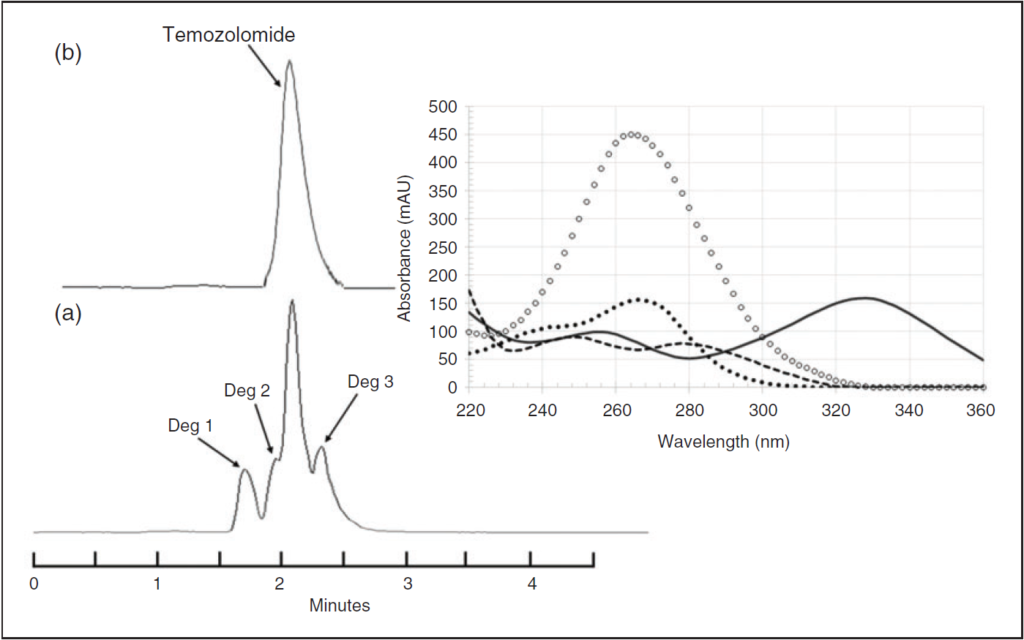
- Dosage de l’irinotécan par HPLC,
- Dosage du SN-38 (produit de dégradation),
- Mesure de pH et l’aspect macroscopique,
- Stérilité microbiologique,
- Méthodes validées selon ICH Q2(R1).
Résultats :
- Concentration en irinotécan stable à ±5 % sur une période de 84 jour,
- SN-38 < 1 % dans toutes les conditions,
- pH stable avec absence d’altération visuelle,
- Stérilité microbiologique conservée y compris après stress thermique,
- Bonne tenue de la solution jusqu’à 7 jours à 30 °C.
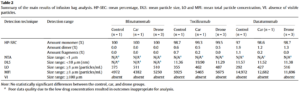
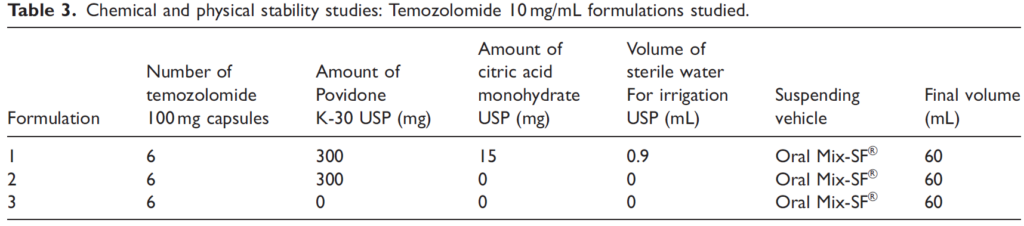
Évolution des concentrations d’irinotécan(exprimées en pourcentage de la concentration initiale à t = 0 h), du pH, de l’osmolalité, de la luminance (L*) et de la chromaticité (a*, b*) dans des poches contenant 200 mg, 300 mg et 370 mg d’irinotécan lors de l’étude de stabilité simulée en condition d’utilisation à température ambiante (25 °C). Après 84 jours de conservation à 4 °C, la poche de perfusion a été placée à 25 °C et analysée aux temps suivants : 0h, 5h, 8h, 10h et 24h.
| Paramètre | Stabilité à long terme (84 j à 4°C) | Stabilité en usage simulé (24 h à 25°C) |
| Concentration (HPLC) | ≥ 95% concentration initiale. | Variation ≤ ±5% (pour les concentrations 200,300 et 370 mg) |
| Produits de dégradation | Aucun pic détecté. | Aucun pic détecté. |
| Largeur de pic (w) | Stable (ex : 0,274±0,005 pour 200 mg). | Stable (ex: 0,277±0,004 pour 200 mg). |
| Aspect visuel | Limpide, sans particules. | Aucun changement. |
| Turbidité (OD) | ≤ 0,005 ± 0,002 | ≤ 0,006 ± 0,001. |
| Chromaticité (L*a*b*) | Stable (ex: à 200 mg : L*=99,89±0,05) | Valeurs identiques au temps 0. |
| pH | Stable (3,71–3,95 selon dose) | Stable (3,61–3,90 selon dose) |
| Osmolalité | Stable (281–282 mOsm/kg). | Stable (280–284 mOsm/kg). |
| Stérilité microbiologique | Maintenue (J0 à J84). | Maintenue. |
Points forts :
- Méthodologie complète (stabilité physique, chimique et microbiologie),
- Validation de la stabilité en conditions d’utilisation simulées.
Points faibles :
- Absence d’évaluation de la photosensibilité,
- Étude limitée à trois dosages,
- Pas de comparaison avec d’autres matériaux de conditionnement.
Conclusion/Implications en pratique :
La stabilité à long terme de l’irinotécan à des doses arrondies normalisées sélectionnées (200 mg – 300 mg – 370 mg) dans des poches de polyoléfine NaCl 0,9 % a été confirmée pendant au moins 84 jours à 4 °C et dans l’obscurité d’un point de vue physico-chimique et microbiologique. Au cours de l’étude simulée en cours d’utilisation, aucun signe d’instabilité chimique n’a été observé. En l’absence de données sur la stabilité à la lumière, une protection systématique contre la lumière reste recommandée.
Rédigé par Imane MEHALAINE et Baptiste FULBERT
D'après Andre C, et al. Long term and simulated in-use stabilities of irinotecan chemotherapy polyolefin infusion bags in dose banding conditions, J Oncol Pharm Pract 2025 (in press), disponible en ligne sur https://doi.org/10.1177/10781552251338763